密度泛函理论(Density Functional Theory, DFT)是一种基于电子密度描述多电子体系基态性质的量子力学方法。它将复杂的多电子问题简化为单电子方程的求解,通过电子密度而非波函数来描述体系的电子结构。
我们的DFT计算服务采用先进的交换关联泛函和基组,结合高性能计算资源,为科研人员提供从几何优化、电子结构分析到各种物理化学性质预测的全方位解决方案,助力新材料开发和基础科学研究。
提供全方位的密度泛函理论计算解决方案,满足不同研究需求
寻找分子和晶体的稳定结构,计算平衡几何构型、键长、键角等结构参数,预测最稳定的异构体。
研究物质的电子结构特征,分析电荷密度分布、电子态密度、能带结构等关键电子性质。
研究化学反应路径,计算反应能垒和反应能,预测反应速率和选择性,揭示反应机理。
预测分子和材料的各种光谱性质,辅助实验光谱解析和新型光学材料设计。
研究材料的磁学性质,计算磁矩、交换耦合常数等参数,预测磁性材料的居里温度。
计算物质的热力学性质,包括焓、熵、吉布斯自由能等,预测反应的热力学可行性。
广泛应用于多个学科领域,助力科研创新与技术突破
研究半导体材料的电子结构和光学性质,指导新型半导体器件设计
设计高效能源存储与转换材料,如电池电极材料、催化剂等
研究磁性材料的磁学性质,开发高性能永磁材料和自旋电子学材料
研究催化反应机理,设计高效催化剂,优化催化反应过程
研究纳米材料的结构与性能关系,开发新型纳米功能材料
设计新型光学材料,如发光材料、非线性光学材料等
研究污染物的降解机理,开发环境友好材料和治理技术
研究药物分子的结构与活性关系,辅助药物分子设计与优化
专业团队与先进技术结合,提供高质量的密度泛函理论计算服务

采用经过验证的交换关联泛函和基组,结合高级电子相关校正方法,确保计算结果的高精度和可靠性。
采用CP2K、Quantum ESPRESSO、Gaussian、ORCA等国际主流量子化学计算软件,确保计算的专业性和高效性。
拥有大规模高性能计算集群,能够处理包含上千原子的复杂体系,完成高精度的DFT计算任务。
不仅提供原始计算数据,还进行深入的结果分析,生成专业的分析报告和高质量可视化图像,助力论文发表。
根据客户的具体研究需求,提供个性化的计算方案设计,从方法选择、参数设置到结果分析,全程专业指导。
标准化的服务流程,确保计算质量与效率
深入了解您的研究目标和具体需求,结合专业知识提供最优计算方案,明确计算体系、方法、参数和预期结果。
根据研究需求构建合理的初始模型,包括分子结构搭建、晶体结构建模、表面模型构建等,准备计算输入文件。
根据研究体系和性质选择合适的交换关联泛函、基组和计算参数,进行测试计算以验证方法的适用性。
在高性能计算集群上执行DFT计算,监控计算过程,确保计算顺利完成,处理可能出现的技术问题。
对计算结果进行全面分析,提取关键物理化学参数,生成专业的分析报告和高质量的可视化图像。
提供计算结果解读和专业咨询,根据需求进行计算方案优化和补充计算,助力研究成果发表。
采用国际主流的量子化学计算软件,确保计算结果的可靠性
运行最快的开源第一性原理材料计算和模拟软件
通用量子化学软件包
开源量子 espresso 软件包
量子化学计算程序
周期性密度泛函理论程序
阿姆斯特丹密度泛函程序
材料科学模拟软件
开源量子化学软件
已为众多高校和科研机构提供专业的密度泛函理论计算服务
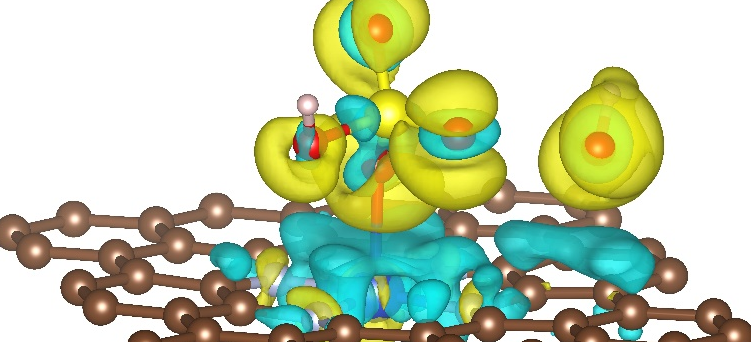
为某高校催化团队设计高效CO₂还原催化剂,通过DFT计算预测活性位点和反应路径,指导实验合成
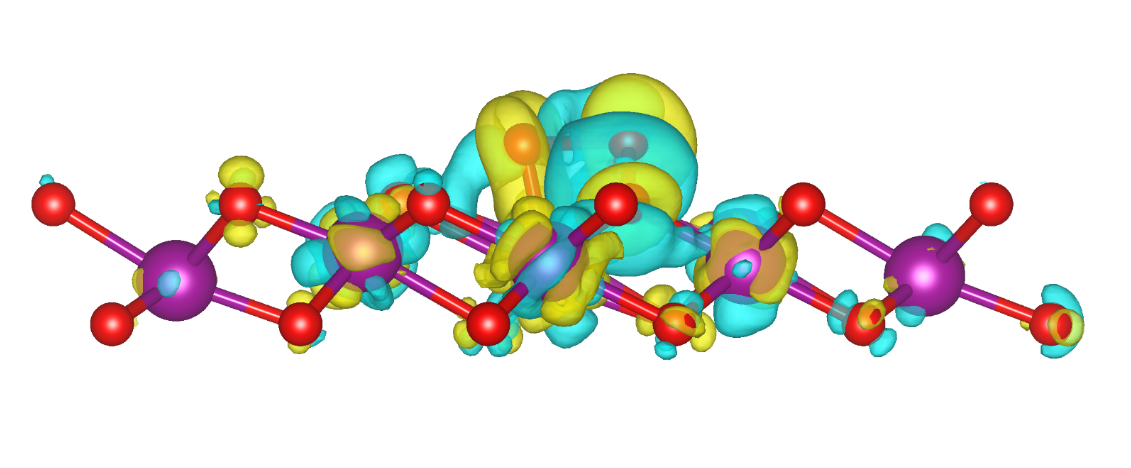
为某研究所计算新型二维材料的电子结构和输运性质,预测其在电子器件中的应用潜力
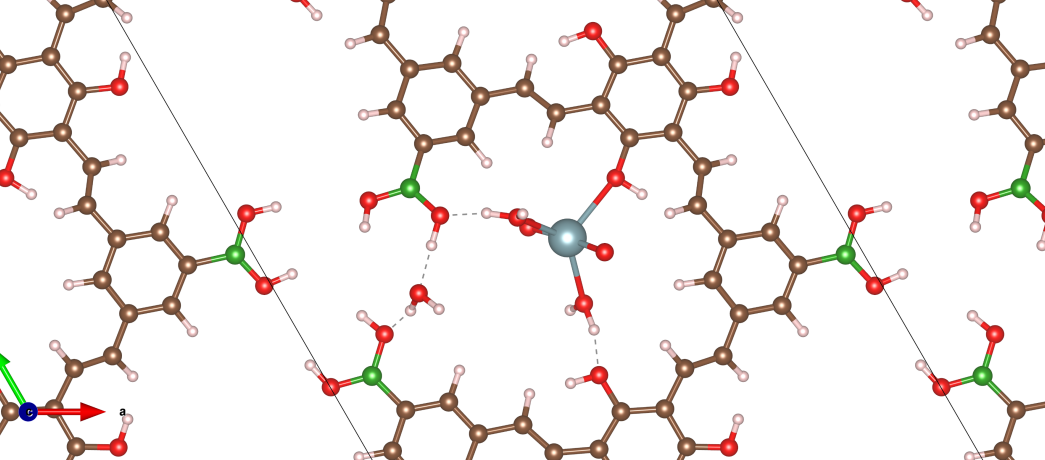
为某能源公司计算锂离子电池电极材料的电化学性能,预测其容量和循环稳定性,指导材料筛选
计算系列稀土永磁材料的交换耦合常数和居里温度,研究成分-结构-磁性关系,指导高性能磁体设计
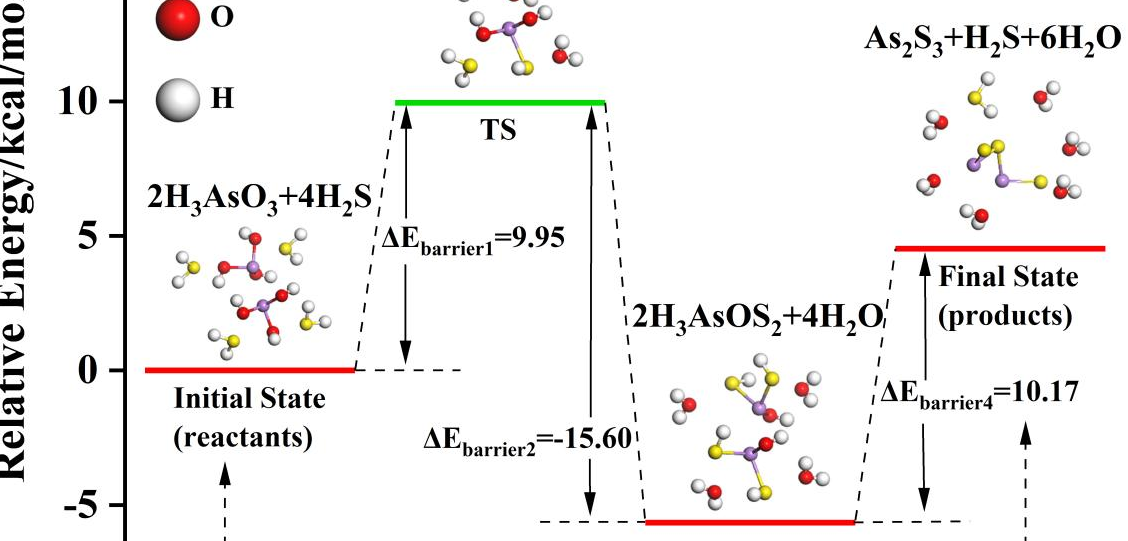
研究抗肿瘤药物分子与靶点蛋白的相互作用机理,计算结合能和反应路径,辅助药物分子优化
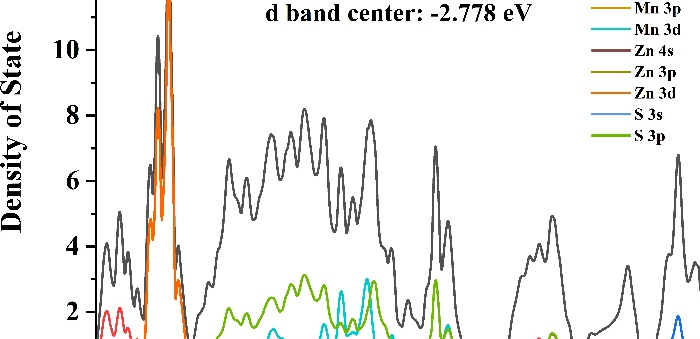
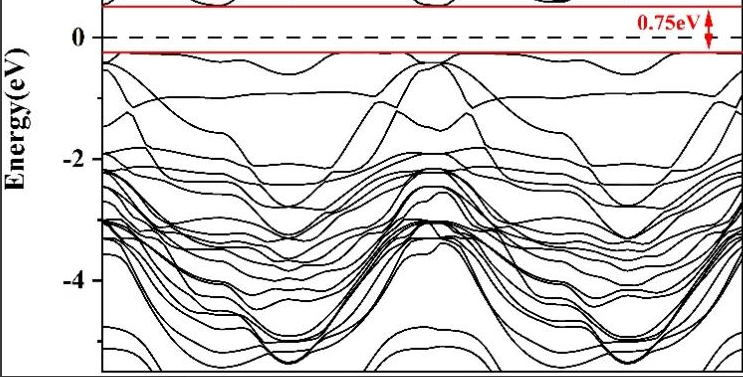
计算新型半导体材料的能带结构和光学吸收谱,预测其在光电器件中的应用性能
关于DFT计算服务的常见疑问解答
无论您有任何疑问或需求,都可以通过以下方式联系我们,专业顾问将为您提供一对一服务
kaiser1204@163.com
199-3892-9951
微信号:JZS999_18
四川省成都市金牛区金府路593号7栋12层9号
