




九章算科技
免费获取计算方案
一站式科研服务
夏日特惠
预存返现超值优享
专业计算模拟平台
免费机会
咨询即送一对一专业指导
论文致谢
最高奖励5000元现金红包
预存好礼
最高返15%
邀请返现
组队双方均获返现
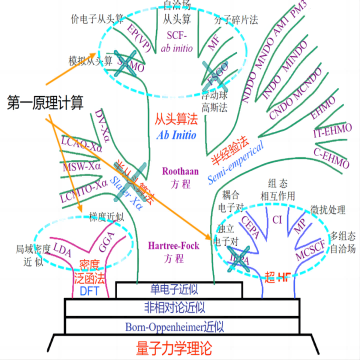
第一性原理
计算价格:
定制方案
预约次数:
4354次
服务周期:
平均10-15个工作日
99.8%
满意度
项目介绍
第一性原理计算的基本思想是将多个原子构成的体系看成是由多个电子和原子核组成的系统,并根据量子力学的基本原理对问题进行最大限度的“非经验性”处理。它只需要5个基本常数(m0,e,h,c,kB)就可以计算出体系的能量和电子结构等物理性质。它可以确定已知材料的结构和基础性质,并实现原子级别的精准控制,是现阶段解决实验理论问题和预测新材料结构性能的有力工具。并且,第一性原理计算不需要开展真实的实验,极大地节省了实验成本,现已被广泛应用于化学、物理、生命科学和材料学等领域。
适合的研究方向包括但不限于:催化、电池、半导体、金属材料、非金属材料、合金、纳米材料等
可以计算的体系包括但不限于:晶体、非晶、二维材料、表面、界面、固体等
可以计算的内容包括但不限于:
材料的几何结构参数(如键长、键角、二面角、晶格常数、原子位置等)
材料的电子结构信息(如电荷密度、电荷差分密度、态密度、能带、费米能级、功函数、ELF等)
材料的光学性质(如介电常数等)
材料的力学性质(如弹性模量等)
材料的磁学性质(如磁导率等)
材料的晶格动力学性质(如声子谱等)
材料的表面性质(如吸附能,催化计算等)
复合材料的性质(异质结等内容)等等。
相关项目
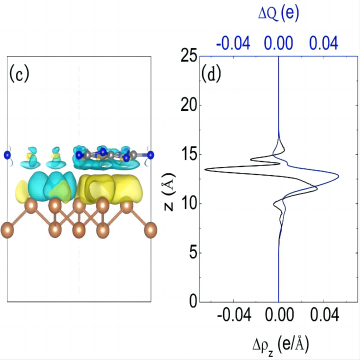
差分电荷
差分电荷计算是一种计算化学方法,用于确定分子中原子之间的电荷分布。它可以提供有关分子的电子密度、键的极性以及分子间相互作用的信息。差分电荷计算的基本思想是通过计算分子中原子的电子数目和电负性来确定原子的电荷。常见的差分电荷计算方法包括:自然键轨道分析(Natural Bond Orbital, NBO)、Mulliken电荷分析、 稳定性电荷分析(Charge Stability Analysis, CSA)。这些方法在计算原子的电荷时采用了不同的近似和假设,因此得到的结果可能会有一定的差异。选择适合特定研究目的的方法,并结合实验数据进行验证和解释,可以获得更准确和可靠的电荷分布信息。
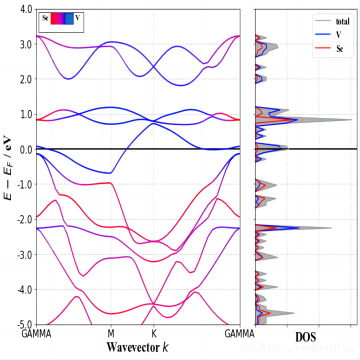
态密度
态密度(Density of States,DOS)是固体物理学和凝聚态物理学中的一个重要概念,用于描述在能量空间中的电子或量子态的分布情况。态密度提供了描述材料中电子能级分布的信息,对于理解材料的电子结构和性质具有重要意义。态密度可以分为两种类型:电子态密度和晶格振动态密度。电子态密度(Electronic Density of States,EDOS)、晶格振动态密度(Phonon Density of States,PDOS)、态密度可以通过实验测量或理论计算得到。实验上,常用的技术包括光电子能谱(Photoemission Spectroscopy,PES)、扫描隧道显微镜(Scanning Tunneling Microscopy,STM)和中子散射等。理论计算方法包括密度泛函理论(Density Functional Theory,DFT)和格林函数方法等。
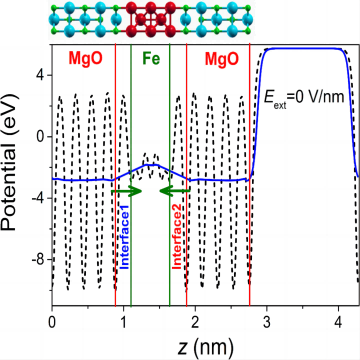
费米能级
费米能级(Fermi level)是固体物理学中的一个重要概念,用于描述在零温下填充电子能级的情况。费米能级可以理解为在零温下,填充电子能级的最高能量。根据泡利不相容原理(Pauli Exclusion Principle),每个电子态只能容纳一个电子,且不同电子态的电子自旋方向必须相反。在填充电子能级时,从低能级开始,逐渐向高能级填充电子,直到填满所有可用的电子态。
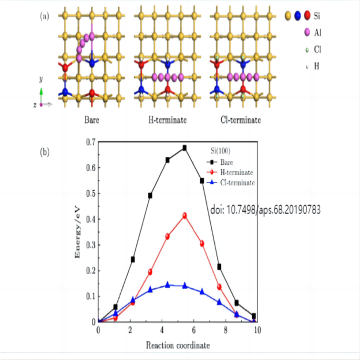
迁移能垒
迁移能垒(Migration energy barrier)是指在固体中原子或分子从一个位置迁移到另一个位置所需克服的能垒。它是描述原子或分子在固体中扩散、迁移或移动的过程中所需的能量障碍。 在固体中,原子或分子的迁移通常涉及克服位能垒或势垒,这是由于相邻原子或分子之间的相互作用力造成的。当原子或分子试图跨越势垒时,需要提供足够的能量以克服这个能垒才能进行迁移。迁移能垒的大小决定了原子或分子在固体中扩散的速率和性质。
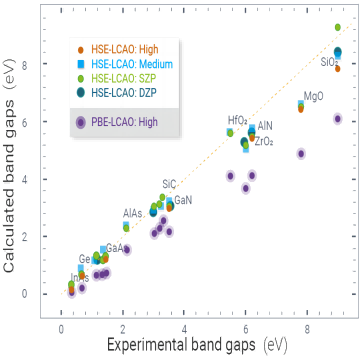
杂化泛函(HSE06)
杂化泛函(Hybrid functional)是一种在密度泛函理论(Density Functional Theory,DFT)框架下用于计算电子结构的方法。它是传统的局域密度近似(Local Density Approximation,LDA)和广义梯度近似(Generalized Gradient Approximation,GGA)的扩展,通过引入一定程度的非局域交换能修正来改善DFT在描述电子相关性和能带结构方面的准确性。在DFT中,电子的总能量可以通过电子密度的泛函表达。传统的LDA和GGA近似中,交换-相关能泛函主要依赖于电子的局域密度或梯度。然而,这些近似方法对于描述电子相关性和能带结构的一些重要现象,如金属-绝缘体过渡、带隙大小和电子态的定性预测等,可能存在一定的不足。
客服顾问

扫码咨询详情
专业顾问为您答疑解惑
客服热线

199-3892-9951
推荐项目
分子动力学